Publications
in reversed chronological order.
2022
-
 Insights into the structural stability of major groove RNA triplexes by WAXS-guided MD simulationsYen-Lin Chen, Weiwei He, Serdal Kirmizialtin, and 1 more authorCell Reports Physical Science, 2022
Insights into the structural stability of major groove RNA triplexes by WAXS-guided MD simulationsYen-Lin Chen, Weiwei He, Serdal Kirmizialtin, and 1 more authorCell Reports Physical Science, 2022RNA triple helices are commonly observed tertiary motifs that are associated with critical biological functions, including signal transduction. Because the recognition of their biological importance is relatively recent, their full range of structural properties has not yet been elucidated. The integration of solution wide-angle X-ray scattering (WAXS) with molecular dynamics (MD) simulations, described here, provides a new way to capture the structures of major-groove RNA triplexes that evade crystallographic characterization. This method yields excellent agreement between measured and computed WAXS profiles and allows for an atomically detailed visualization of these motifs. Using correlation maps, the relationship between well-defined features in the scattering profiles and real space characteristics of RNA molecules is defined, including the subtle conformational variations in the double-stranded RNA upon the incorporation of a third strand by base triples. This readily applicable approach has the potential to provide insight into interactions that stabilize RNA tertiary structure that enables function.

2021
-
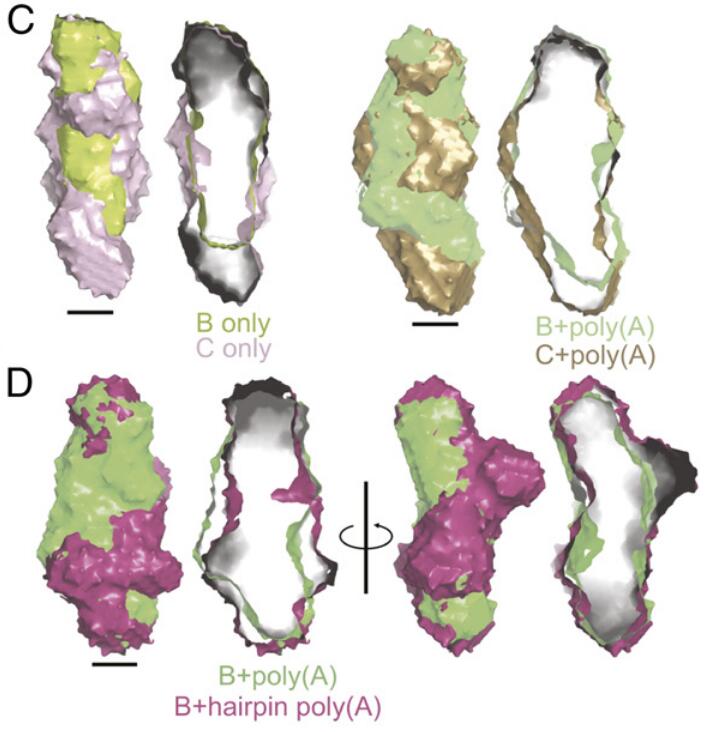 Structural analyses of an RNA stability element interacting with poly(A)Seyed-Fakhreddin Torabi, Yen-Lin Chen, Kaiming Zhang, and 8 more authorsProceedings of the National Academy of Sciences, 2021
Structural analyses of an RNA stability element interacting with poly(A)Seyed-Fakhreddin Torabi, Yen-Lin Chen, Kaiming Zhang, and 8 more authorsProceedings of the National Academy of Sciences, 2021Cis-acting RNA elements are crucial for the regulation of polyadenylated RNA stability. The element for nuclear expression (ENE) contains a U-rich internal loop flanked by short helices. An ENE stabilizes RNA by sequestering the poly(A) tail via formation of a triplex structure that inhibits a rapid deadenylation-dependent decay pathway. Structure-based bioinformatic studies identified numerous ENE-like elements in evolutionarily diverse genomes, including a subclass containing two ENE motifs separated by a short double-helical region (double ENEs [dENEs]). Here, the structure of a dENE derived from a rice transposable element (TWIFB1) before and after poly(A) binding (∼24 kDa and ∼33 kDa, respectively) is investigated. We combine biochemical structure probing, small angle X-ray scattering (SAXS), and cryo‐electron microscopy (cryo-EM) to investigate the dENE structure and its local and global structural changes upon poly(A) binding. Our data reveal 1) the directionality of poly(A) binding to the dENE, and 2) that the dENE-poly(A) interaction involves a motif that protects the 3ʹ-most seven adenylates of the poly(A). Furthermore, we demonstrate that the dENE does not undergo a dramatic global conformational change upon poly(A) binding. These findings are consistent with the recently solved crystal structure of a dENE+poly(A) complex [S.-F. Torabi et al., Science 371, eabe6523 (2021)]. Identification of additional modes of poly(A)–RNA interaction opens new venues for better understanding of poly(A) tail biology.
-
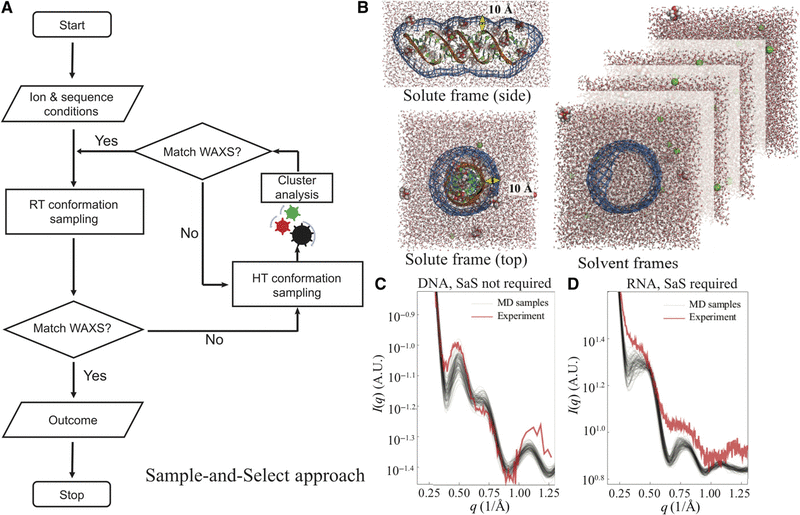 The structural plasticity of nucleic acid duplexes revealed by WAXS and MDWeiwei He, Yen-Lin Chen, Lois Pollack, and 1 more authorScience Advances, 2021
The structural plasticity of nucleic acid duplexes revealed by WAXS and MDWeiwei He, Yen-Lin Chen, Lois Pollack, and 1 more authorScience Advances, 2021The structural diversity of nucleic acid duplexes reflects the critical roles of sequence, ions, and waters. Double-stranded DNA (dsDNA) and RNA (dsRNA) helices display an unusual structural diversity. Some structural variations are linked to sequence and may serve as signaling units for protein-binding partners. Therefore, elucidating the mechanisms and factors that modulate these variations is of fundamental importance. While the structural diversity of dsDNA has been extensively studied, similar studies have not been performed for dsRNA. Because of the increasing awareness of RNA’s diverse biological roles, such studies are timely and increasingly important. We integrate solution x-ray scattering at wide angles (WAXS) with all-atom molecular dynamics simulations to explore the conformational ensemble of duplex topologies for different sequences and salt conditions. These tightly coordinated studies identify robust correlations between features in the WAXS profiles and duplex geometry and enable atomic-level insights into the structural diversity of DNA and RNA duplexes. Notably, dsRNA displays a marked sensitivity to the valence and identity of its associated cations.


2020
-
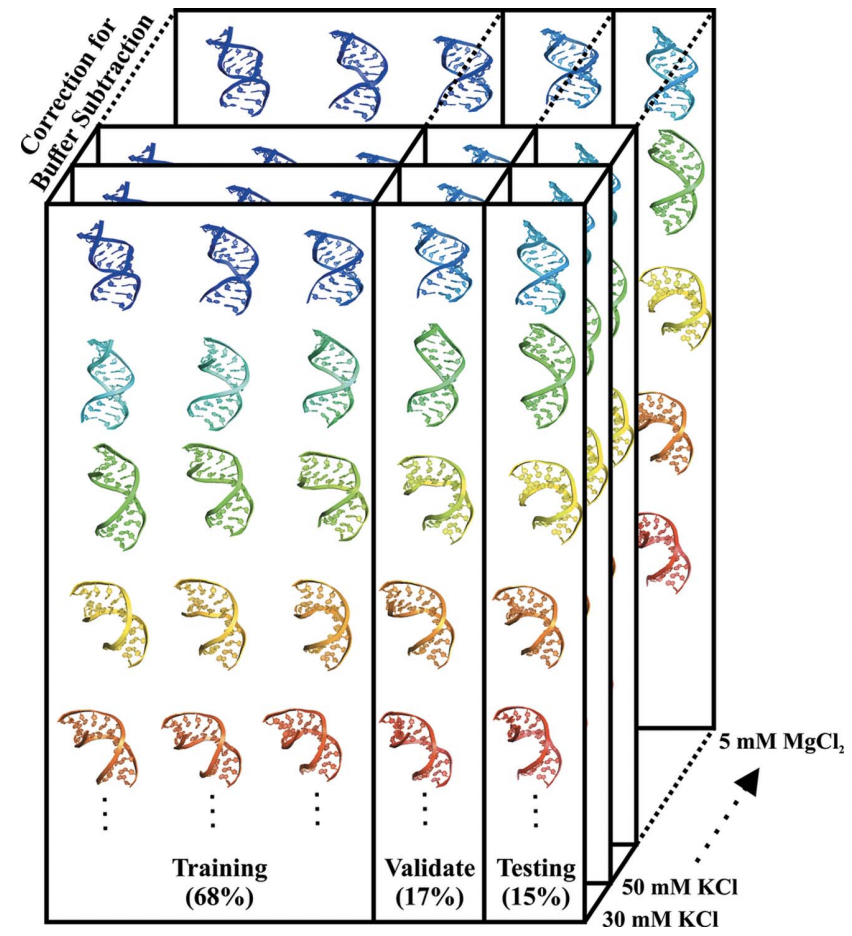 Machine learning deciphers structural features of RNA duplexes measured with solution X-ray scatteringYen-Lin Chen, and Lois PollackIUCrJ, Sep 2020
Machine learning deciphers structural features of RNA duplexes measured with solution X-ray scatteringYen-Lin Chen, and Lois PollackIUCrJ, Sep 2020Macromolecular structures can be determined from solution X-ray scattering. Small-angle X-ray scattering (SAXS) provides global structural information on length scales of 10s to 100s of Ångstroms, and many algorithms are available to convert SAXS data into low-resolution structural envelopes. Extension of measurements to wider scattering angles (WAXS or wide-angle X-ray scattering) can sharpen the resolution to below 10Å, filling in structural details that can be critical for biological function. These WAXS profiles are especially challenging to interpret because of the significant contribution of solvent in addition to solute on these smaller length scales. Based on training with molecular dynamics generated models, the application of extreme gradient boosting (XGBoost) is discussed, which is a supervised machine learning (ML) approach to interpret features in solution scattering profiles. These ML methods are applied to predict key structural parameters of double-stranded ribonucleic acid (dsRNA) duplexes. Duplex conformations vary with salt and sequence and directly impact the foldability of functional RNA molecules. The strong structural periodicities in these duplexes yield scattering profiles with rich sets of features at intermediate-to-wide scattering angles. In the ML models, these profiles are treated as 1D images or features. These ML models identify specific scattering angles, or regions of scattering angles, which correspond with and successfully predict distinct structural parameters. Thus, this work demonstrates that ML strategies can integrate theoretical molecular models with experimental solution scattering data, providing a new framework for extracting highly relevant structural information from solution experiments on biological macromolecules.
-
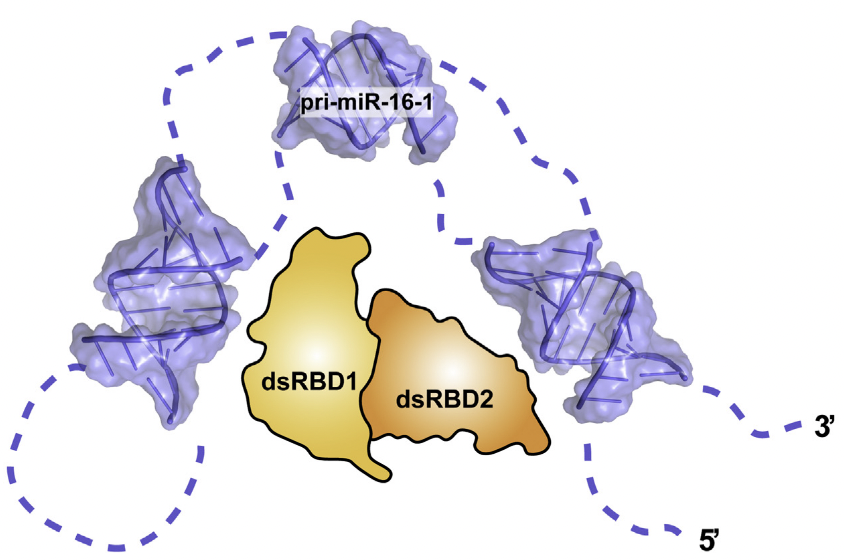 Elucidating the Role of Microprocessor Protein DGCR8 in Bending RNA StructuresSuzette A. Pabit, Yen-Lin Chen, Emery T. Usher, and 3 more authorsBiophysical Journal, Dec 2020
Elucidating the Role of Microprocessor Protein DGCR8 in Bending RNA StructuresSuzette A. Pabit, Yen-Lin Chen, Emery T. Usher, and 3 more authorsBiophysical Journal, Dec 2020Although conformational dynamics of RNA molecules are potentially important in microRNA (miRNA) processing, the role of the protein binding partners in facilitating the requisite structural changes is not well understood. In previous work, we and others have demonstrated that nonduplex structural elements and the conformational flexibility they support are necessary for efficient RNA binding and cleavage by the proteins associated with the two major stages of miRNA processing. However, recent studies showed that the protein DGCR8 binds primary miRNA and duplex RNA with similar affinities. Here, we study RNA binding by a small recombinant construct of the DGCR8 protein and the RNA conformation changes that result. This construct, the DGCR8 core, contains two double-stranded RNA-binding domains (dsRBDs) and a C-terminal tail. To assess conformational changes resulting from binding, we applied small-angle x-ray scattering with contrast variation to detect conformational changes of primary-miR-16-1 in complex with the DGCR8 core. This method reports only on the RNA conformation within the complex and suggests that the protein bends the RNA upon binding. Supporting work using smFRET to study the conformation of RNA duplexes bound to the core also shows bending. Together, these studies elucidate the role of DGCR8 in interacting with RNA during the early stages of miRNA processing.
-
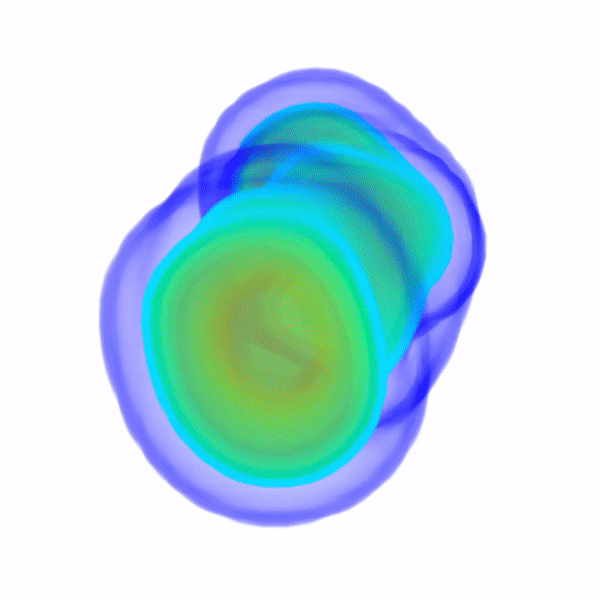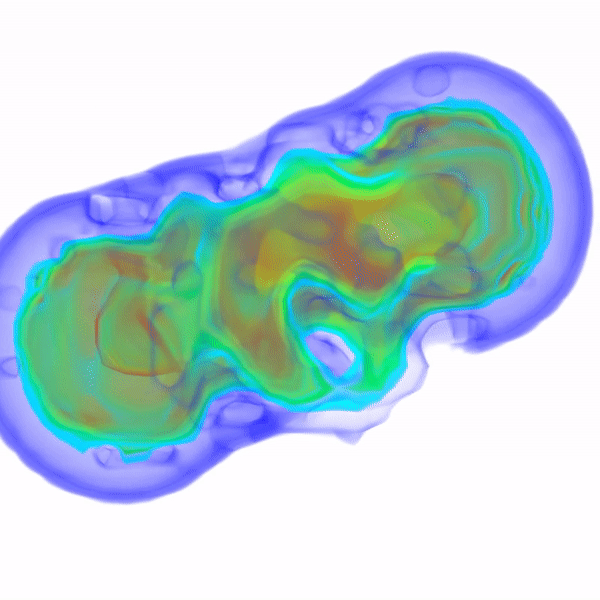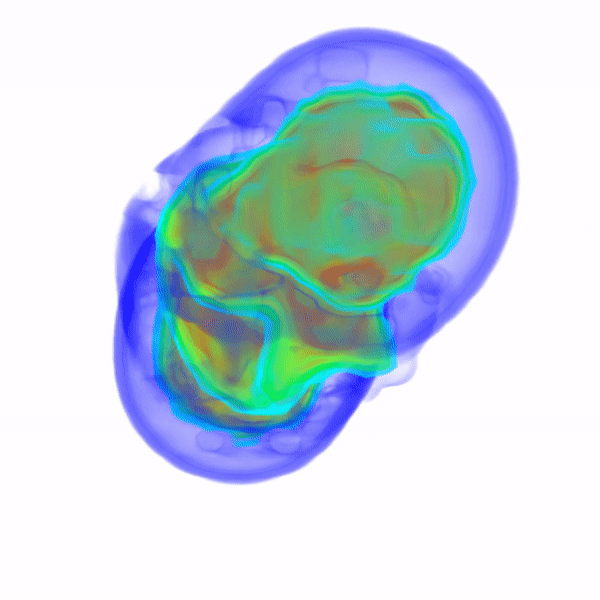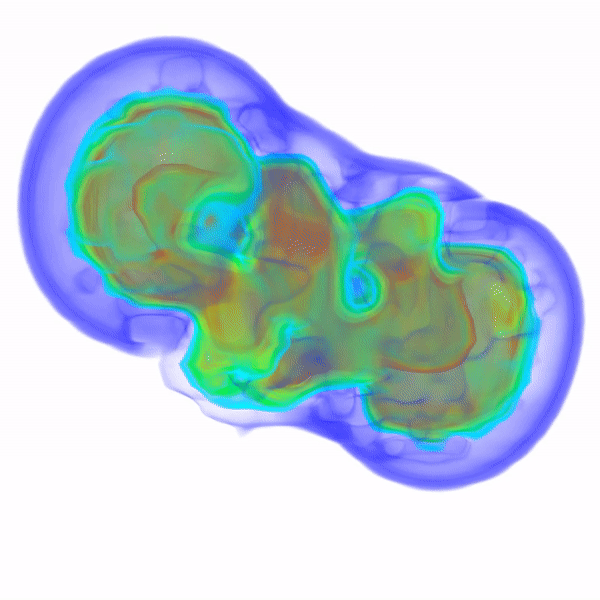 In Vitro Electron Density Refinement from Solution X-ray Scattering in the Wide-Angle RegimeYen-Lin Chen, and Lois PollackarXiv, Dec 2020
In Vitro Electron Density Refinement from Solution X-ray Scattering in the Wide-Angle RegimeYen-Lin Chen, and Lois PollackarXiv, Dec 2020We present Frequency Marching, FM, an algorithm that refines three-dimensional electron density distributions from solution X-ray scattering data in both the small- and wide-angle regimes. This algorithm is based on a series of optimization steps, marching along the frequency (reciprocal) space and refining detailed periodic structures with the corresponding real-space resolution. Buffer subtraction and excluded volumes, key factors in extracting the signatures of the biomolecule of interest from the sample, are accounted for using implicit density models. We provide the numerical and analytical basis of the FM algorithm. We demonstrate this technique by application to structured and unstructured nucleic acid systems, where higher resolution features are carved out of low resolution reconstructions as the algorithm marches into wider angles.



2019
-
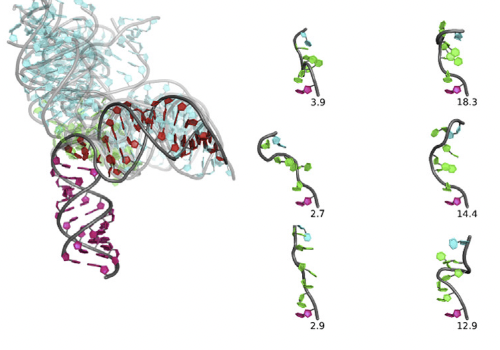 Conformations of an RNA Helix-Junction-Helix Construct Revealed by SAXS Refinement of MD SimulationsYen-Lin Chen, Tongsik Lee, Ron Elber, and 1 more authorBiophysical Journal, Jan 2019
Conformations of an RNA Helix-Junction-Helix Construct Revealed by SAXS Refinement of MD SimulationsYen-Lin Chen, Tongsik Lee, Ron Elber, and 1 more authorBiophysical Journal, Jan 2019RNA is involved in a broad range of biological processes that extend far beyond translation. Many of RNA’s recently discovered functions rely on folding to a specific conformation or transitioning between conformations. The RNA structure contains rigid, short basepaired regions connected by more flexible linkers. Studies of model constructs such as small helix-junction-helix (HJH) motifs are useful in understanding how these elements work together to determine RNA conformation. Here, we reveal the full ensemble of solution structures assumed by a model RNA HJH. We apply small-angle x-ray scattering and an ensemble optimization method to selectively refine models generated by all-atom molecular dynamics simulations. The expectation of a broad distribution of helix orientations, at and above physiological ionic strength, is not met. Instead, this analysis shows that the HJH structures are dominated by two distinct conformations at moderate to high ionic strength. Atomic structures, selected from the molecular dynamics simulations, reveal strong base-base interactions in the junction that critically constrain the conformational space available to the HJH molecule and lead to a surprising re-extension at high salt. These results are corroborated by comparison with previous single-molecule fluorescence resonance energy transfer experiments on the same constructs.
- Salt Dependence of A-Form RNA Duplexes: Structures and ImplicationsYen-Lin Chen, and Lois PollackThe Journal of Physical Chemistry B, Jan 2019PMID: 31638810
The biological functions of RNA range from gene regulation through catalysis and depend critically on its structure and flexibility. Conformational variations of flexible, non-base-paired components, including RNA hinges, bulges, or single-stranded tails, are well documented. Recent work has also identified variations in the structure of ubiquitous, base-paired duplexes found in almost all functional RNAs. Duplexes anchor the structures of folded RNAs, and their surface features are recognized by partner molecules. To date, no consistent picture has been obtained that describes the range of conformations assumed by RNA duplexes. Here, we apply wide angle, solution X-ray scattering (WAXS) to quantify these variations, by sampling length scales characteristic of helical geometries under different solution conditions. To identify the radius, helical rise, twist, and length of dsRNA helices, we exploit molecular dynamics generated structures, explicit solvent models, and ensemble optimization methods. Our results quantify the substantial and salt-dependent deviations of double-stranded (ds) RNA duplexes from the assumed canonical A-form conformation. Recent experiments underscore the need to properly describe the structures of RNA duplexes when interpreting the salt dependence of RNA conformations.

2018
- How the Conformations of an Internal Junction Contribute to Fold an RNA DomainYen-Lin Chen, Julie L. Sutton, and Lois PollackThe Journal of Physical Chemistry B, Jan 2018PMID: 30285445
Like proteins, some RNAs fold to compact structures. We can model functional RNAs as a series of short, rigid, base-paired elements, connected by non-base-paired nucleotides that serve as junctions. These connecting regions bend and twist, facilitating the formation of tertiary contacts that stabilize compact states. Here, we explore the roles of salt and junction sequence in determining the structures of a ubiquitous connector: an asymmetric internal loop. We focus on the J5/5a junction from the widely studied P4–P6 domain of the Tetrahymena ribozyme. Following the addition of magnesium ions to fold P4–P6, this junction bends dramatically, bringing the two halves of the RNA domain together for tertiary contact engagement. Using single-molecule fluorescence resonance energy transfer (smFRET), we examine the role of sequence and salt on model RNA constructs that contain these junction regions. We explore the wild-type J5/5a junction as well as two sequence variants. These junctions display distinct, salt-dependent conformations. Small-angle X-ray scattering (SAXS) measurements verify that these effects persist in the full-length P4–P6 domain. These measurements underscore the importance of junction sequence and interactions with ions in facilitating RNA folding.